Cystisk fibrose er en medfødt metabolsk sykdom. Kroppsvæsker som spytt, bronkialt slim eller bukspyttkjertelsekresjoner er mye tøffere enn vanlig på grunn av genetisk disponering. Konsekvenser inkluderer luftveisproblemer og fordøyelsesbesvær. Cystisk fibrose er ikke kurerbar. Med konsekvent terapi kan sykdomsforløpet imidlertid bremses. Les her hvilke symptomer som forårsaker cystisk fibrose og hvordan du behandler den.

Cystisk fibrose: kort oversikt
- Beskrivelse: arvelig metabolsk sykdom, forårsaker tøff slimdannelse i lungene og andre organer
- symptomer: Luftveisproblemer, irriterende hoste, lungeinfeksjon, manglende trivsel, fordøyelsesbesvær, alvorlig diaré, fet lever, redusert fruktbarhet
- årsaker: Arv av mangelfulle gener som påvirker konsistensen av kroppsvæsker, utbrudd av sykdommen bare hvis begge foreldrene arver et sykt gen (dominerende recessiv arv)
- diagnose: Blodprøve for immunreaktivt trypsin (IRT), pankreatittassosiert protein (PAP), svetteprøve, genetisk test
- behandling: mukolytiske midler, bronkodilaterende midler, inhalasjon, antibiotika for infeksjoner, kortison, CFTR-modulatorer, lungetransplantasjon
- prognose: ikke kurerbar, selvfølgelig sterkt avhengig av alvorlighetsgraden og tidspunktet for diagnosen, forkortet forventet levealder
Cystisk fibrose: beskrivelse
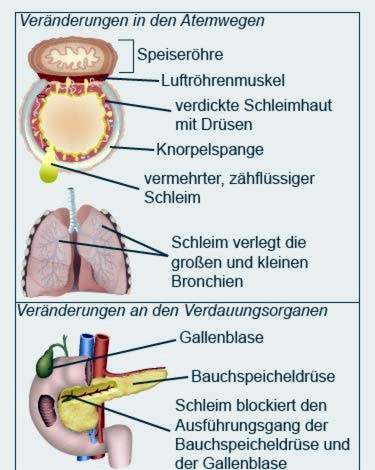
Cystisk fibrose (også kalt cystisk fibrose) er en arvelig metabolsk sykdom. Dannelsen av forskjellige kroppsvæsker forstyrres. Sekresjonene i lungene, bukspyttkjertelen og andre organer er mer tyktflytende enn hos friske mennesker.
Tøft slim
Det tøffe slimet tetter blant annet de små grenene i bronkiene og kanalene i de indre organene. Pusten og fordøyelsen påvirkes spesielt. I løpet av sykdommen kan organene fungere verre og verre.
Feil i arvestoffet
Årsaken til sykdommen er defekter i arvestoffet. Cystisk fibrose er derfor ikke herdbar. Tidspunkt for diagnose og alvorlighetsgrad av symptomer kan variere mye individuelt. Hos mange barn gjør cystisk fibrose en massiv innvirkning fra fødselen, i andre tilfeller gjenkjennes det senere.
Cystisk fibrose: symptomer
Cystisk fibrose symptomer kan variere veldig fra pasient til pasient. Sykdommen påvirker funksjonen til forskjellige organer, men spesielt lungene og fordøyelsessystemet.
Gjenkjenne tidlige tegn
De første symptomene på cystisk fibrose varierer individuelt. I de fleste tilfeller vises cystisk fibrose symptomer i løpet av det første leveåret. Dermed kan sykdommen vanligvis diagnostiseres tidlig og startes raskt med en terapi. Noen pasienter har imidlertid betydelige klager bare i ungdomstiden. Ikke alle berørte personer viser hele spekteret av mulige symptomer. Alvorligheten av symptomene varierer også.
Forandrede kroppsvæsker
Ved cystisk fibrose forstyrres dannelsen av de såkalte kloridionkanalene i cellene. Dette endrer sammensetningen av kroppsvæsker. Den enkleste måten å oppdage denne endringen i svetten hos de berørte. Svetten din er saltere enn sunne mennesker. Saltene natrium og klorid, som tilhører de såkalte elektrolytter, er beriket i svetten. Som et resultat av svette mister pasienter som lider av cystisk fibrose i økende grad kroppssalter.
Varierte symptomer på cystisk fibrose
Sykdommen påvirker en lang rekke organsystemer. Ofte vises de første symptomene på cystisk fibrose i lungene og fordøyelseskanalen. I løpet av livet kan ytterligere klager legges til. Gjennom målrettet terapi kan symptomene behandles godt. Imidlertid kan symptomene også nå truende proporsjoner. Det er spesielt farlig når bronkiene tetter seg gjennom det viskøse slimet. Da kan pasientene kveles i ekstreme tilfeller.
Cystisk fibrose: symptomer på lungen
Pusteproblemer og irriterende hoste
I de fleste tilfeller forekommer symptomer på lungene bare ved litt eldre spedbarn ved cystisk fibrose. Nyfødte har vanligvis ingen pusteproblemer. Cystisk fibrose-symptomer kommer ofte til uttrykk i form av en hoste-halslignende, kronisk, irriterende hoste hos litt eldre barn. Slimet i luftveiene er økt, seigt og tyktflytende. Dette hindrer luftstrømmen i lungene. Over tid utvikler en progressiv luftveisnød.
Hyppige infeksjoner
Økt slimproduksjon i lungene gjør det lettere for bakterier å kolonisere og forårsake infeksjon. Gjentagende lungebetennelse eller bronkialinfeksjoner er hovedsakelig forårsaket av bakterier som stafylokokker og Pseudomonas-arter. Den forstyrrede saltbalansen i lungene hindrer også kroppens forsvar. Også lungeblødninger kan forekomme. Et typisk tegn på dette er hoste av blodblandet slim.
Selv om lungene er skadet fra en tidlig alder, er de første symptomene på cystisk fibrose i luftveiene ofte bare i barneskolealder eller til og med senere. Symptomene forekommer noen ganger bare når store deler av lungevevet allerede er ødelagt eller luftveiene er kraftig innsnevret.
Cystisk fibrose: symptomer på bukspyttkjertelen
Hos pasienter med cystisk fibrose blir bukspyttkjertelen ofte betent. Den utskiller en sekresjon som inneholder blant annet enzymer for fordøyelse av fett og sukker. Hos pasienter med cystisk fibrose kondenserer sekresjonen på grunn av dens viskositet tilbake og forårsaker betennelse.
Når prosessen skrider frem, blir bukspyttkjertelen vev og herdet. Legene snakker om fibrose. Fibrosen ødelegger gradvis bukspyttkjertelen. I tillegg til gallen danner bukspyttkjertelen også insulin, som er nødvendig, blant annet for sukkerutnyttelsen i kroppen. Pasienter som er litt eldre (fra ungdomstiden) utvikler ofte diabetes mellitus.
Cystisk fibrose: symptomer på galle
Bukspyttkjertelen og galleblæren har en felles kanal i tarmen. Derfor kan tilbakestrømningen av bukspyttkjertelsekret også forårsake betennelse i galleblæren. Ofte dannes gallestein, som fullstendig kan blokkere galleblæren fra galleblæren.
Cystisk fibrose: symptomer på fordøyelseskanalen
I tillegg til klager på lungene, påvirker cystisk fibrose hovedsakelig fordøyelsen. På grunn av mangelen på galle er for eksempel fettfordøyelsen nedsatt. Ofte tåler pasienter fet mat dårlig. Den inntatte maten skilles stort sett ut ufordøyd. Typiske er da veldig omfangsrike og myke avføring.
Diaré og vekstlidelser
Berørte barn, inkludert spedbarn, lider ofte av alvorlig diaré. Selv om de drikker og spiser godt, øker de knapt. Vekstlidelser og underernæring er derfor ytterligere klassiske konsekvenser av sykdommen.
Slike klager innen fordøyelsen kan imidlertid også forekomme ved andre sykdommer. Bare i kombinasjon med luftveisproblemer er de derfor en karakteristisk indikasjon på cystisk fibrose, som absolutt bør undersøkes.
anal prolaps
I det videre løpet kan cystisk fibrose forårsake forskjellige komplikasjoner i fordøyelseskanalen. Det vanligste er en såkalt anal prolaps. Ved anal prolaps bukker tarmslimhinnen ut av anus. En slik hendelse må behandles kirurgisk så snart som mulig.
tarmobstruksjon
Involvering av tarmen (invaginasjon) eller tarmobstruksjon (ileus) er også vanlig. Begge komplikasjonene er assosiert med sterke magesmerter og betydelige fordøyelsesproblemer. Smertene oppstår vanligvis i spurts, spesielt etter å ha spist. En tarmhindring er dødelig hvis den ikke blir behandlet. Krampaktig, akutt magesmerter bør derfor alltid avklares av lege.
Cystisk fibrose: symptomer på leveren
fettlever
Under tilbakestrømningen av galle lider også leveren. Hos mange pasienter utvikler fet lever i løpet av sykdommen. Tretthet, tap av matlyst, oppblåsthet og flatulens, og i sjeldne tilfeller kan følelser av trykk eller smerter i øvre del av magen oppstå.
levercirrhose
I sjeldne tilfeller utvikler det seg en krympende lever (skrumplever) der leveren er sterkt forstyrret i sin funksjon. Den manifesterer seg først i form av gulsott (gulsott). Tegn på gulsott er den gulaktige misfargingen av hvitt i øynene. Etter lang tid oppstår også hjerteproblemer og ytelsen til de berørte fortsetter å avta.
Cystisk fibrose: redusert fruktbarhet
Mer enn halvparten av alle mannlige pasienter er ufruktbare. Selv om de i de fleste tilfeller kan danne fruktbar sæd, kan de ikke komme gjennom vas deferensene fordi de er blokkert av tyktflytende slim.
Berørte kvinner er vanligvis mindre fruktbare. Generelt sett kan de motta og levere et barn. Imidlertid akkumuleres det i deres eggleder tøft slim, som sædcellene knapt kan trenge gjennom. Spesielt i en høyere alder avtar sannsynligheten for graviditet raskt.
Cystisk fibrose: symptomer hos barn
Cystisk fibrose er en genetisk sykdom. Den har alltid vært til stede siden fødselen. Men de klassiske symptomene på cystisk fibrose forekommer ikke alltid i barndommen. Imidlertid er det ofte allerede uspesifikke instruksjoner som bør følges. Dette gjelder spesielt hvis tilfeller av cystisk fibrose allerede har oppstått i familien.
Oppblåst mage
En indikasjon på at det er en metabolsk lidelse er for eksempel en oppblåst mage i lang tid. Ofte lider barna av diaré. Hos nyfødte kan en markert forsinket første avføring (Kindspech) være en indikasjon på cystisk fibrose. I mange tilfeller oppstår vekst- og vekstforstyrrelser selv om barna spiser med sug. Bare i sjeldne tilfeller fører det også til forstoppelse (forstoppelse) som et resultat av sykdommen.
Skrallende pust
Andre symptomer som kan indikere cystisk fibrose inkluderer skramlende pust og alvorlig rastløshet. Mange barn lider av kronisk betennelse i bihulene. Disse er hovedsakelig merket av ikke akkurat lokaliserbare smerter i ansiktet. Nasale polypper er mer vanlig hos barn med cystisk fibrose enn hos friske barn.
Hvis barn lider av pusteproblemer eller fordøyelsesbesvær lenger, bør lege alltid konsulteres som et forsiktighetsregler. Hos barn kan livstruende situasjoner raskt oppstå fordi de ikke selv kan artikulere klagene sine eller deres alvorlighetsgrad ikke kan vurdere.
Cystisk fibrose: årsaker og risikofaktorer
Cystisk fibrose er forårsaket av en genetisk defekt. Den patologiske forandringen ligger på det syvende kromosomet i det såkalte CFTR-genet.
CFTR-genet (cystisk fibrose-transmembranregulatorgen) inneholder konstruksjonsmanualen for en kanal som kloridioner kommer inn i cellene gjennom. De mangelfulle kloridionkanalene blokkerer transport av salt til visse kroppsceller hos cystisk fibrosepasienter.
De berørte kjertelcellene, men i stedet for den ellers flytende sekresjonen, tøft slim fra. I lungene danner paranasale bihuler, bukspyttkjertelen, i tarmen, i galleveiene og i gonadene så tøffe slimutskillelser med høy saltholdighet.
Cystisk fibrose: hvor truet er mitt barn?
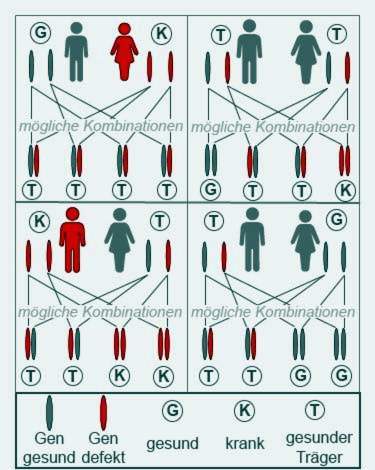
Cystisk fibrose bryter bare ut når begge foreldrene gir et patologisk endret gen til barnet sitt. Disse foreldrene er da stort sett begge selv sunne, men bærere av genet.
Personer med cystisk fibrose er bare delvis fruktbare. Noen pasienter blir fortsatt foreldre. Syke fedre eller mødre viderefører imidlertid alltid et sjukt gen, da begge CFTR-genene har informasjon om cystisk fibrose. Barna deres blir imidlertid bare syke hvis de også får et sykt gen fra den andre forelderen.
Par hvis familier allerede har hatt cystisk fibrose, bør søke genetisk rådgivning før de planlegger et svangerskap.
Preimplantasjonsdiagnostikk
Foreldre som kan føde cystisk fibrose kan gjennomgå pre-implantasjon genetisk diagnose. Ved genetisk testing av preimplantasjon blir oocyttene kunstig befruktet. De første celledelingene finner sted i prøverøret (in vitro).
Før det settes inn et embryo, testes det for endrede genegenskaper. Bare embryoer blir implantert som ikke har det syke genet. Uavhengig av dette kan det også under graviditet undersøkes om barnet vil utvikle CF senere.
Cystisk fibrose: undersøkelser og diagnose
I motsetning til for noen år siden, gjennomgår de fleste sykehus i dag rutinert neonatal screening. Dette inkluderer studier på cystisk fibrose.
Screening i blod og svette
For screeningen tas blod fra den nyfødte. Cystisk fibrose-test involverer flere stadier:
blodprøve
Test for forhøyede nivåer av immunreaktivt trypsin (IRT) og pankreatittassosiert protein (PAP). I tilfelle avvik, finner svetteprøven sted.
sveising test
Cystisk fibrose pasienter har en betydelig høyere saltholdighet hos svette enn friske mennesker. For svetteprøve av cystisk fibrose måles innholdet av saltene natrium og klorid i kroppens svette. Hos barn blir svetten på underarmen samlet og deretter analysert. Oppstår det mistanke her, vil en genetisk test bli utført.
gentest
Hos pasienter med cystisk fibrose har det såkalte CFTR-genet, som gir planen for spesifikke ionekanaler, endret seg. Denne konstruksjonshåndboken er lang, den består av omtrent 6500 basepar. Overalt kan en feil krype inn i kode, men feilene har forskjellige effekter. Derfor testes bare de vanligste avvikene i koden.
Familiehistorie gir hint
Hvis det ikke er utført noen passende nyfødtscreening og mistenkt cystisk fibrose kommer opp senere, er familielegen eller internisten den rette kontakten. I en innledende samtale registrerer denne personen sykehistorien (anamnese). Ved mistanke om cystisk fibrose, er spesiell oppmerksomhet rettet til familiehistorien.
Fysisk undersøkelse
Deretter foregår en fysisk undersøkelse. Legen lytter til lungene og skanner de indre organene. Han kan allerede utelukke noen andre tilstander som er assosiert med symptomer som ligner på cystisk fibrose.
I tillegg kan røntgenundersøkelser vise blokkering av luftveiene og lungene. Laboratorieundersøkelser gir indikasjoner på funksjonelle begrensninger i de indre organene. Selv hos voksne som er mistenkt for cystisk fibrose, gir en svetteprøve viktige bevis for diagnosen.
Testing av familiemedlemmer
Hvis cystisk fibrose finnes i en familie, er det fornuftig at alle andre familiemedlemmer også gjennomgår en undersøkelse. Cystisk fibrose kan også forekomme i svekkede former. Det tar ofte mange flere år før cystisk fibrose manifesterer seg med klare symptomer. Likevel er tidlig diagnose og cystisk fibrose terapi også viktig for de som blir berørt for å øke forventet levealder.
Cystisk fibrose: behandling
Cystisk fibrose er ikke kurerbar. Barn født med cystisk fibrose lider av effekten av sykdommen i løpet av livet. En kombinasjon av fysioterapi, medisiner og inhalasjoner kan imidlertid redusere sykdommens progresjon. En cystisk fibrose-behandling bør derfor først og fremst oppnå at de berørte kan føre et så normalt liv som mulig.
Lære å leve med sykdommen
Spesielt for barn er det viktig å lære å takle sykdommen på lang sikt. Barn med cystisk fibrose bør lære så tidlig som mulig hva sykdommen betyr og hvordan den påvirker kroppen. Her kan et sykehusopphold med spesielle treningsenheter være nyttig. I prosessen lærer barn og foreldre å mate seg selv, hvordan sport ser ut og hvordan de oppfører seg best i kritiske situasjoner.
Hjelp for lungene
Det er forskjellige alternativer for å behandle symptomene. Avhengig av pasientens alder og alvorlighetsgraden av symptomene, anbefales forskjellige tilnærminger.
Mukolytiske midler
Cystisk fibrose pasienter lider mest av lungeproblemer. Ved regelmessig innånding med spesielle tilsetningsstoffer (mukolytika) løses det viskøse slim opp og kan lett hostes av.
Bronkiale dilatasjonsmidler
Såkalte beta-2-sympatomimetika øker også bronkiene, noe som i tillegg letter pusten.
Antibiotika mot bakterier
På grunn av dårlig ventilasjon i lungene lider personer med cystisk fibrose av bakterieinfeksjoner i luftveiene mye oftere. På den annen side hjelper antibiotika som gis i god tid. I noen tilfeller er det fornuftig å inhalere disse permanent.
Antiinflammatoriske medisiner
Hos mange pasienter er luftveiene ofte eller kronisk betente. Da hjelper antiinflammatoriske midler som kortison.
CFTR modulatorer
I mellomtiden er de første medisinene utviklet som forbedrer den nedsatte funksjonen til ionekanalene. Imidlertid fungerer de bare for visse mutasjoner og dermed bare for en liten del av pasientene. Deres effektivitet er også begrenset. Forbedring av medisinene er intensivt undersøkt. For første gang ville de begynne med årsaken til sykdommen i stedet for bare symptomene.
Lungetransplantasjon – det siste håpet
I alvorlige tilfeller er en lungetransplantasjon også et alternativ. Så drastisk som dette trinnet ser ut til å begynne med, kan mange pasienter føre et betydelig lavere belastning etterpå.
Mat riktig i cystisk fibrose
Siden cystisk fibrose også forstyrrer fordøyelsen, må pasientene følge nøye med kostholdet. Du bør foretrekke et kosthold med mye protein og karbohydrater.
Det finnes også vitamintilskudd og mineraler. Sistnevnte erstatter saltene som svetter pasientene i store mengder. Siden bukspyttkjertelen ikke fungerer som de skal, får barn fordøyelsesenzymer medisiner som et supplement til måltider.
Opplev vaksinasjoner
Vaksinasjon er spesielt viktig for pasienter med CF. Hos dem har bakterier enklere lek, og de blir ofte mer alvorlige enn pasienter som ikke er forhåndslastet. Spesielt vaksiner mot meslinger og pneumokokker anbefales. I tillegg skal hvert år være en influensavaksine.
Cystisk fibrose: sykdomsforløp og prognose
Cystisk fibrose er forårsaket av en endring i genomet og er derfor ikke kurerbar. For personer med cystisk fibrose er forventet levealder og livskvalitet vanligvis betydelig redusert. Uten behandling forverres helsetilstanden raskt, og de berørte lever vanligvis ikke lenge.
Med rettidig og konsistent terapi kan sykdomsforløpet reduseres betydelig. I mellomtiden lever pasienter mye lenger enn for noen år siden. Gjennomsnittlig forventet levealder for cystisk fibrose er for tiden rundt 40 år. Men mange lever også med sykdommen i 50 år og mer.
Komplikasjoner og følger
Selv med intensiv terapi kan komplikasjoner oppstå igjen og igjen ved cystisk fibrose. Oftest er det akutt luftveisproblem på grunn av dårlig lungeventilasjon. Individuelle områder i lungen kan til og med kollapse (alektase).
Ofte utvikles kronisk bronkitt eller lungebetennelse. Sopp kan også påvirke lungene.
I tillegg kan skift i væske- og elektrolyttbalansen utløse sjokk og sirkulasjonssvikt.
Andre komplikasjoner og følgere inkluderer:
- kronisk leversykdom, spesielt skrumplever
- Galleblæren betennelser og gallestein
- kronisk betennelse i bukspyttkjertelen
- forstyrret hjertefunksjon
- akutt tarmobstruksjon (ileus)
- Intestinal invaginasjon
- underernæring
- Diabetes mellitus
- Begrenset fruktbarhet hos kvinner, eller infertilitet hos mannlige pasienter
Cystisk fibrose er en arvelig sykdom, forebygging er derfor ikke mulig. Personer med risiko for familiær risiko bør søke genetisk rådgivning hvis de ønsker å få barn. En genetisk test blir utført og det blir undersøkt om CFTR-genet er endret. Avhengig av om en eller begge partnere bærer genet, kan risikoen for avkommet beregnes.
I mellomtiden er genetisk diagnose (PID) også mulig i cystisk fibrose preimplantasjon i Tyskland. Forutsetningen for dette er alltid godkjenning av et etisk utvalg. Oocytter blir befruktet utenfor livmoren og bare embryoer uten de problematiske cystiske fibrose-generene blir brukt.
Ytterligere informasjon
retningslinjer:
- S2 Consensus Guideline «Diagnosis of Cystic Fibrosis» fra Society for Pediatric Pulmonology (2013)
- S3 retningslinje «Lungesykdom ved cystisk fibrose» av Society for Pediatric Pulmonology (2013)
Støtte grupper:
- Cystic Fibrosis e.V.